
Identifying the correct mechanism of a reaction can still be one of the toughest tasks facing chemists. For example, when designing a catalyst,
May 1, 2017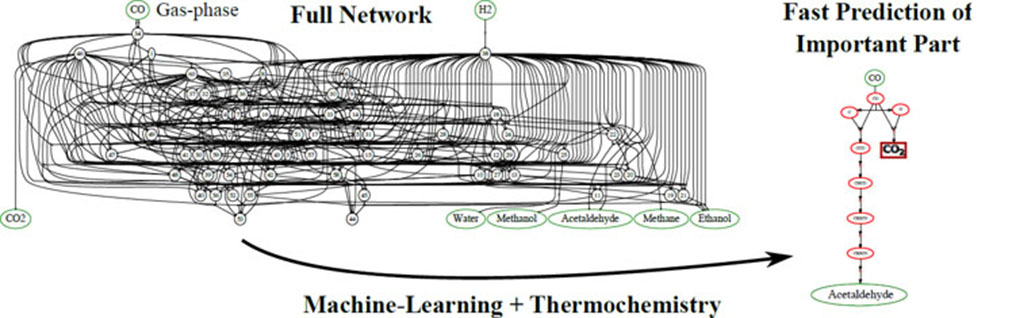
The Science
Turning fossil resources or biomass into fuels and countless other chemical reaction networks often involves hundreds of different species and reactions. The networks result in thousands of potential pathways for a reaction to proceed. A new computational technique uses machine-learning techniques and thermochemistry data to readily eliminate unlikely paths. Further, the technique reveals a subset of the most important pathways. More intensive computational methods can then be applied to only the important pathways.
The Impact
Decreasing the number of intensive calculations needed to identify reaction paths saves both researcher time and expensive high-performance computational resources. In addition, focusing the computation on key pathways improves the accuracy of the reaction network models. This new technique benefits all fields of chemistry that involve large reaction networks. Specifically, the technique benefits catalysis and combustion.
Summary
Identifying the correct mechanism of a reaction can still be one of the toughest tasks facing chemists. For example, when designing a catalyst, researchers must know which mechanism will occur in order to achieve the desired product. Additionally, the researcher will seek to minimize both undesirable side-products and the energy required to carry out the reaction. However, even a seemingly simple reaction can have thousands of possible pathways to reach a specific product. Fortunately, in most cases, only small portions of the possible pathways are relevant to the reaction. These important pathways are known as the rate-limiting steps. By focusing theoretical simulation efforts initially on identifying these important rate-limiting paths, researchers have reduced the time and computing resources needed to perform detailed investigations of very large reaction networks. This new technique uses approximations based on thermochemical and kinetic energy data to exclude clearly unfavorable sections of the reaction network. Intensive computational calculations are then applied to the remaining pathways, which represent the rate-limiting elementary reactions. Excluding the unnecessary sections of the reaction network allows the rate-limiting pathways to be studied in detail but now requires a significantly smaller number of intense calculations be carried out on high-performance computers, thus saving a great deal of researcher time and computer resources. Specifically, the researchers, from the SUNCAT Center for Interface Science and Catalysis and the Georgia Institute of Technology, applied this method to the reaction of syngas (carbon monoxide and hydrogen) on a rhodium(111) surface, a reaction scientists are developing to convert fossil and biomass resources to useful chemicals and fuels. Using the described method, the team determined the most likely reaction mechanism used far fewer intense calculations than previously possible. This technique is broadly applicable to virtually any reaction network and could improve computational methods for all fields of chemistry.
Contact
Jens K. Nørskov
SUNCAT Institute for Interface Science and Catalysis
Stanford University, Stanford Linear Accelerator
norskov@stanford.edu
Funding
Funding was provided by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences to the SUNCAT Center for Interface Science and Catalysis. Additionally, A.J.M. was supported by the Department of Defense through the National Defense Science & Engineering Graduate Fellowship Program.
Publications
Z.W. Ulissi, A.J. Medford, T. Bligaard, and J.K. Nørskov, "To address surface reaction network complexity using scaling relations machine learning and DFT calculations ." Nature Communications 8, 14621 (2017). [DOI: 10.1038/ncomms14621]
." Nature Communications 8, 14621 (2017). [DOI: 10.1038/ncomms14621]
Related Links
SUNCAT Center for Interface Science and Catalysis
Highlight Categories
Performer/Facility: University, DOE Laboratory
Additional: Collaborations, Non-DOE Interagency Collaboration