
Local scaling self-interaction correction (LSIC) overcomes long-standing problems for accurate simulation of chemical reactions from start to finish.
March 20, 2020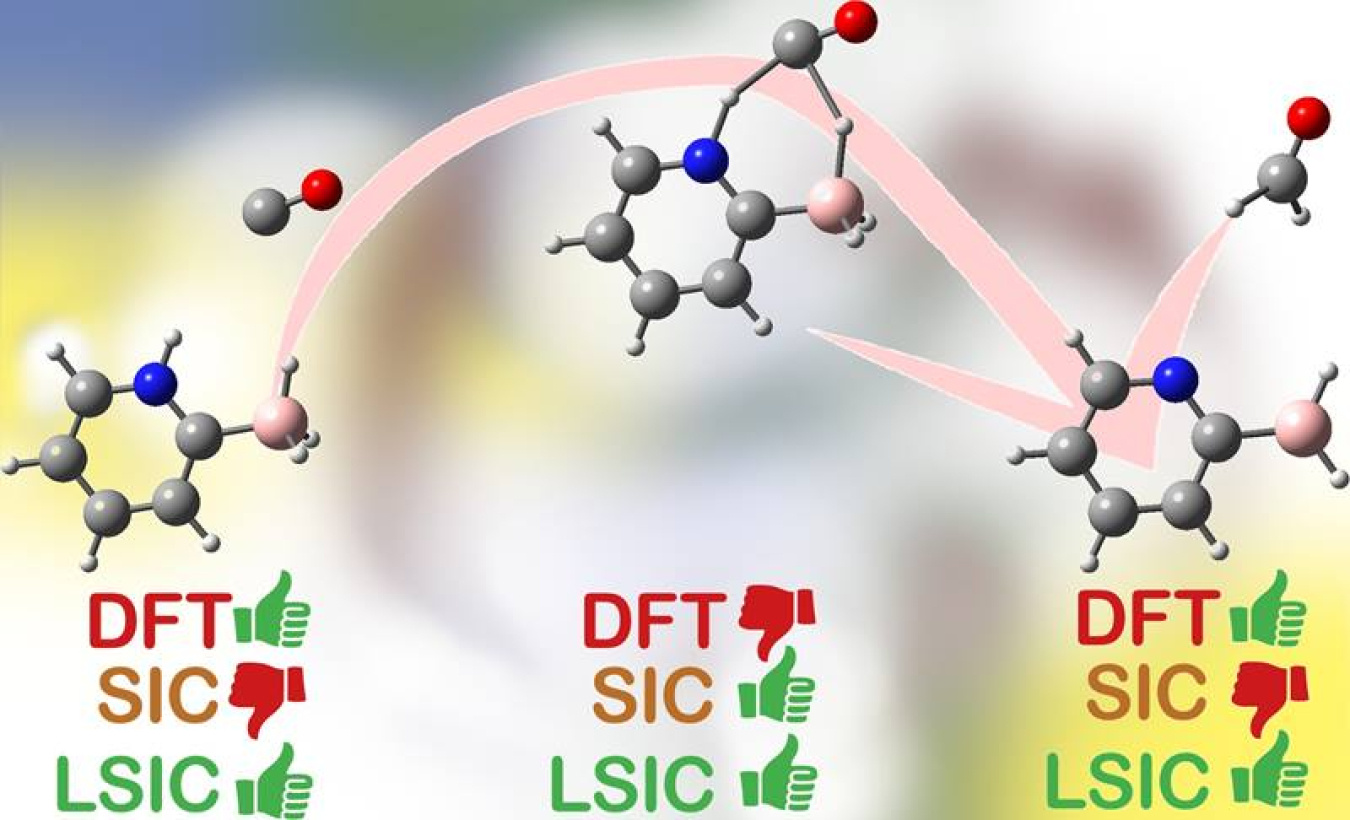
The Science
Nearly all computer models of molecules and materials are based on density functional theory (DFT) approximations. These methods help efficiently simulate the structure, energy, and properties of atoms in molecules or materials. DFT approximations often lead to what researchers call self-interaction error of the electrons. Scientists have several methods for correcting self-interaction error. These methods work well for some chemical arrangements. But they introduce new errors in other arrangements. Scientists have now created a new method, local scaling self-interaction correction (LSIC). This new method removes self-interaction errors without hurting accuracy. It works over the entire span of chemical arrangements.
The Impact
The new LSIC method increases the accuracy of DFT methods. Specifically, it enables scientists to better model atoms during the entire span of chemical reactions. These reactions start with the chemical bonds between the atoms of a molecule or solid at or near their equilibrium positions. Reactions then pass through a phase when chemical bonds are being stretched or broken. The reaction completes with the new arrangement of atoms and bonds between them again at or near equilibrium. By describing all these phases of the chemical reaction accurately, the new LSIC method will allow scientists to more reliably model chemical transformations than possible with current methods. The new method has potential applications in a broad range of chemistry and materials science research.
Summary
Scientists use DFT-based methods to study molecules and solids because these methods often deliver high accuracy at low computational cost. The efficiency of DFT arises from approximating the exact quantum mechanics of complex many-electron systems. However, these approximations inevitably result in spurious electron self-interaction that is too large to be ignored in some situations. This means DFT methods more reliably predict properties accurately when atoms are near stable positions because self-interaction errors tend to become more pronounced when molecules and solids are not in equilibrium, such as when chemical bonds are stretched or broken during chemical reactions. Scientists have previously used methods to correct for self-interaction errors and improve the accuracy in these situations, but applying SIC methods comes at the cost of less accurate descriptions of molecules under equilibrium conditions. This paradox means that a single method cannot be used to follow a chemical reaction from a stable starting point, through a transition state, to a stable end point. The new LSIC method resolves this paradox by giving accurate results in all conditions. LSIC works by selectively removing self-interaction errors in spatial regions where it is needed and gives no correction where the electron density is uniform and DFT approximations are exact.
Contact
Rajendra R. Zope
Physics Department, University of Texas at El Paso
rrzope@utep.edu
Funding
DOE Office of Science, Office of Basic Energy Sciences, Computational Chemical Sciences Program
Publications
Zope, R.R., et al., “A step in the direction of resolving the paradox of Perdew-Zunger self-interaction correction.” J. Chem. Phys. 151, 214108 (2019). [DOI: 10.1063/1.5129533]