
Scientists develop a new tool to find viruses in complex genomic data sets.
Biological and Environmental Research
January 30, 2020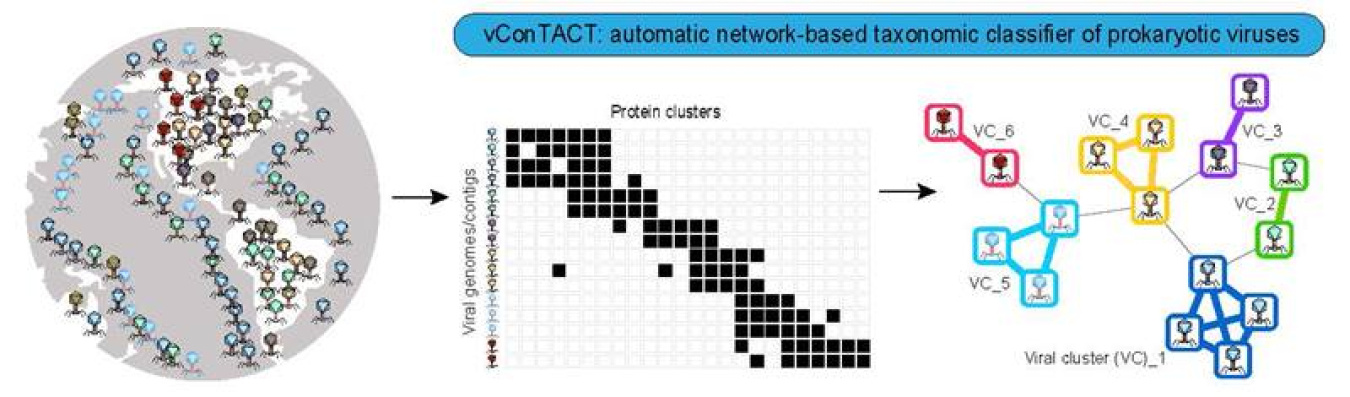
The Science
Researchers developed open-source software that can classify viruses in ways that previous tools could not. Scientists have limited data on viruses that they cannot grow in laboratories. That lack of information makes them especially hard to classify. The new system uses viral genes to separate out viruses that are difficult to distinguish from each other into distinct groups. This separation is a key step in organizing and isolating viruses that scientists are particularly interested in. Tests using information from known viruses have shown the new software is very accurate.
The Impact
Research on viruses is an important frontier in environmental science. In fact, viruses that invade bacteria and archaea are most likely critical to all ecosystems. Every environment contains a myriad of viruses that scientists cannot grow in the laboratory. However, the lack of a framework that can classify large numbers of viruses and includes viruses’ relationship with their hosts holds back progress in this area. This software tool provides a new standard for classifying viruses that scientists have detected in DNA from field and other environmental samples.
Summary
Classification of environmental viruses, specifically uncultivated viral genomes (‘UVIGS’) is a key step to organizing the virosphere and isolating viral groups of potential interest. Single-gene or full genome phylogenies are commonly used to classify viruses within a known framework of virus classification. However, a high rate of gene exchange in and between bacterial viruses (‘phages’) makes it difficult to classify highly divergent phages with the limited data available. A team of researchers developed vConTACT 2.0, an open source, community-available, network-based software application to establish prokaryotic virus taxonomy that scales to thousands of uncultivated virus genomes/fragments, while integrating multiple confidence scores for all taxonomic predictions. Performance tests show the predictions of the new software with currently classified viruses to be very accurate (International Committee on Taxonomy of Viruses; >91% genus-level assignments at 97% accuracy). This approach can also resolve highly recombinogenic taxa through an integrated distance-based hierarchical approach, and remaining discrepancies likely will require changes to current viral taxonomy guides. vConTACT 2.0 also automatically classified 1,364 previously unclassified reference viruses. The software application can be scaled to modern metagenomic datasets with a robust reference network and could potentially uncover thousands more viral sequences. Together, these efforts provide a systematic reference network and a robust, scalable taxonomic analysis tool that is critically needed for the research community.
Contact
Matthew Sullivan
Ohio State University
sullivan.948@osu.edu
Jennifer Pett-Ridge
Lawrence Livermore National Laboratory
pettridge2@llnl.gov
PM Contact
Dawn Adin
DOE Office of Biological and Environmental Research, Biological Systems Science Division
dawn.adin@science.doe.gov
Ramana Madupu
DOE Office of Biological and Environmental Research, Biological Systems Science Division
ramana.madupu@science.doe.gov
Funding
Funding was provided in part by the Department of Energy’s Genome Sciences Program Soil Microbiome Scientific Focus Area award to Lawrence Livermore National Laboratory, NSF Biological Oceanography awards, and a Gordon and Betty Moore Foundation Investigator Award to M.B.S. Funding was provided to J.R.B. by the Intramural Research Program of the National Institutes of Health (NIH) National Library of Medicine. The work conducted by the US Department of Energy Joint Genome Institute is supported by the Office of Science of the US Department of Energy. This work was also funded in part through Battelle Memorial Institute’s prime contract with the US National Institute of Allergy and Infectious Diseases (NIAID).
Publications
Jang, H.B., Bolduc, B., Zablocki, O., Kuhn, J.H., Roux, S., Adriaenssens, E.M., Brister, J.R., Kropinski, A.M., Krupovic, M., Lavigne, R., Turner, D., and Sullivan, M, “Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks.” Nature Biotechnology, 37: 632–639 (2019). [DOI: 10.1038/s41587-019-0100-8]