
Scientists found that gas-phase oxo-exchange of the positively charged protactinium ion, PaO2+, with water was substantially
March 31, 2016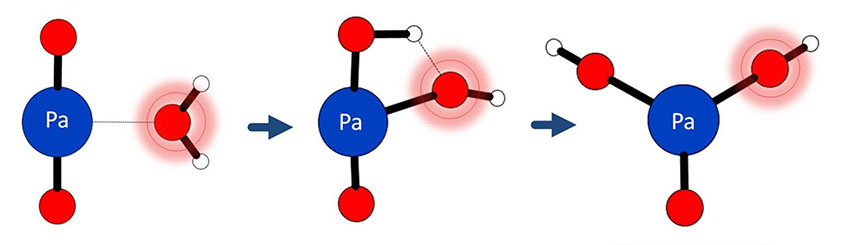
The Science
Found naturally only in trace quantities and seminal to alternate nuclear reactor fuels, protactinium is an extremely rare element that could reveal new trends among nearby actinide elements, including uranium. Scientists at Lawrence Berkeley National Laboratory and Argonne National Laboratory demonstrated that a positively charged protactinium dioxide ion may not exist in aqueous solution like other highly charged actinides, such as uranium and plutonium. Why? Because the energy of hydrating, or adding a water molecule, is very near to hydrolyzing or causing that water molecule to break apart. This is in contrast to the behavior of heavier actinides, such as uranium and plutonium.
The Impact
The molecular-level results provide key fundamental insights into the distinctively different chemistries of protactinium and the other actinides in aqueous solutions. The chemistry of protactinium, which is very difficult to study due to it scarcity and complex chemistry, is important for understanding and controlling its behavior for application in nuclear reactors, as well as for comprehending the overall variable and complex chemistry of the actinide series and transition elements used in everyday technologies.
Summary
Scientists found that gas-phase oxo-exchange of the positively charged protactinium ion, PaO2+, with water was substantially faster than that of its uranium counterpart, UO2+, indicating that the Pa-O bonds are more susceptible to activation and formation of the bis-hydroxide intermediate, PaO(OH)2+. To elucidate the nature of the water adduct of PaO2+, hydration of PaO2+ and UO2+, as well as collision-induced dissociation and ligand-exchange of the water adducts of PaO2+ and UO2+, were studied in an ion trap. The results indicate that, in contrast to UO2(H2O)+, the protactinium hydroxide isomer, PaO(OH)2+, is produced as a gas-phase species close in energy to the hydrate isomer, PaO2(H2O)+. Similar collision-induced dissociation behavior to Th(OH)3+ subports the assignment as PaO(OH)2+. The gas-phase results are consistent with the spontaneous hydrolysis of PaO2+ in aqueous solution, this in contrast to heavier actinide dioxides of uranium, neptunium, and plutonium; the heavier actinide dioxides form stable hydrates in both solution and gas phase. The researchers concluded that the stabilities of oxo-hydroxides relative to oxide hydrates decrease in the order: Th(IV) > Pa(V) > U(V) > Np(V) > Pu(V). This trend suggests increasing covalency, and decreasing ionicity, of actinide oxygen bonds upon proceeding across the actinide series, a key chemical attribute.
Contact
John K. Gibson
Lawrence Berkeley National Laboratory
JKGibson@lbl.gov
Funding
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Heavy Element Chemistry, at Lawrence Berkeley National Laboratory under Contract No. DE-AC02-05CH11231 (P.D.D. and J.K.G.) and by DOE Office of Science, Office of Basic Energy Sciences, Early Career Research Award Program, under Contract DE-AC02-06CH11357 (R.E.W.).
Publications
P. D. Dau, R. E. Wilson, and J. K. Gibson, "Elucidating protactinium hydrolysis: The relative stabilities of PaO2(H2O)+ and PaO(OH)2+ ." Inorganic Chemistry 54, 7474 (2015). [DOI: 10.1021/acs.inorgchem.5b01078].
." Inorganic Chemistry 54, 7474 (2015). [DOI: 10.1021/acs.inorgchem.5b01078].
Related Links
Peculiar Protactinium Nature Article
Highlight Categories
Performer/Facility: DOE Laboratory