
Name: Jonas Kaufman; Institution: University of California, Santa Barbara; Program: DOE Computational Science Graduate Fellowship (DOE CSGF); Education Level: Graduate Student
March 14, 2023In Jonas Kaufman’s research, small things have big effects. The computational materials science doctoral candidate studies how subtle atomic-level changes affect large-scale properties. He says his projects at the University of California, Santa Barbara, “build an understanding of that. I think that’s really cool.”

Kaufman, a Department of Energy Computational Science Graduate Fellowship (DOE CSGF) recipient, developed that appreciation over years. He took high school mathematics and chemistry in Princeton, New Jersey, and went to California to study engineering at Harvey Mudd College. As he learned more about physics, however, he gravitated toward the subject and joined professor Lori Bassman’s group.
Bassman had just begun exploring density functional theory (DFT), a demanding technique to calculate crystals’ electronic structures, including their sub-microscopic quantum mechanical effects.
“I wasn’t really familiar with DFT or even the idea of computational materials science,” which designs and characterizes compounds at the atomic and molecular scales, Kaufman says. DFT expert Aurora Pribram-Jones – a Harvey Mudd and DOE CSGF alumna – advised the group. The result: Kaufman and Pribram-Jones, now a professor at the University of California, Merced, were among authors on a 2017 paper in Physical Review B.
Kaufman’s DFT experience underlies his graduate research with Anton van Der Ven. The group performs calculations from first principles – fundamental physics – to predict materials’ stability and transitions between phases (distinct arrangements of matter). Kaufman focuses on battery electrode compounds, particularly sodium- and potassium-ion substances that could replace standard lithium-ion batteries in certain applications.
Their main tool is Clusters Approach to Statistical Mechanics (CASM), software that uses approximations based on clusters of atoms to predict a material’s macroscopic qualities. The group first uses DFT to calculate energies and other properties of many small structures. Those data are used to train CASM approximations via machine learning. The resulting models run quickly and in large numbers, using a Monte Carlo method that runs simulations based on randomly sampled conditions to predict the system’s attributes.
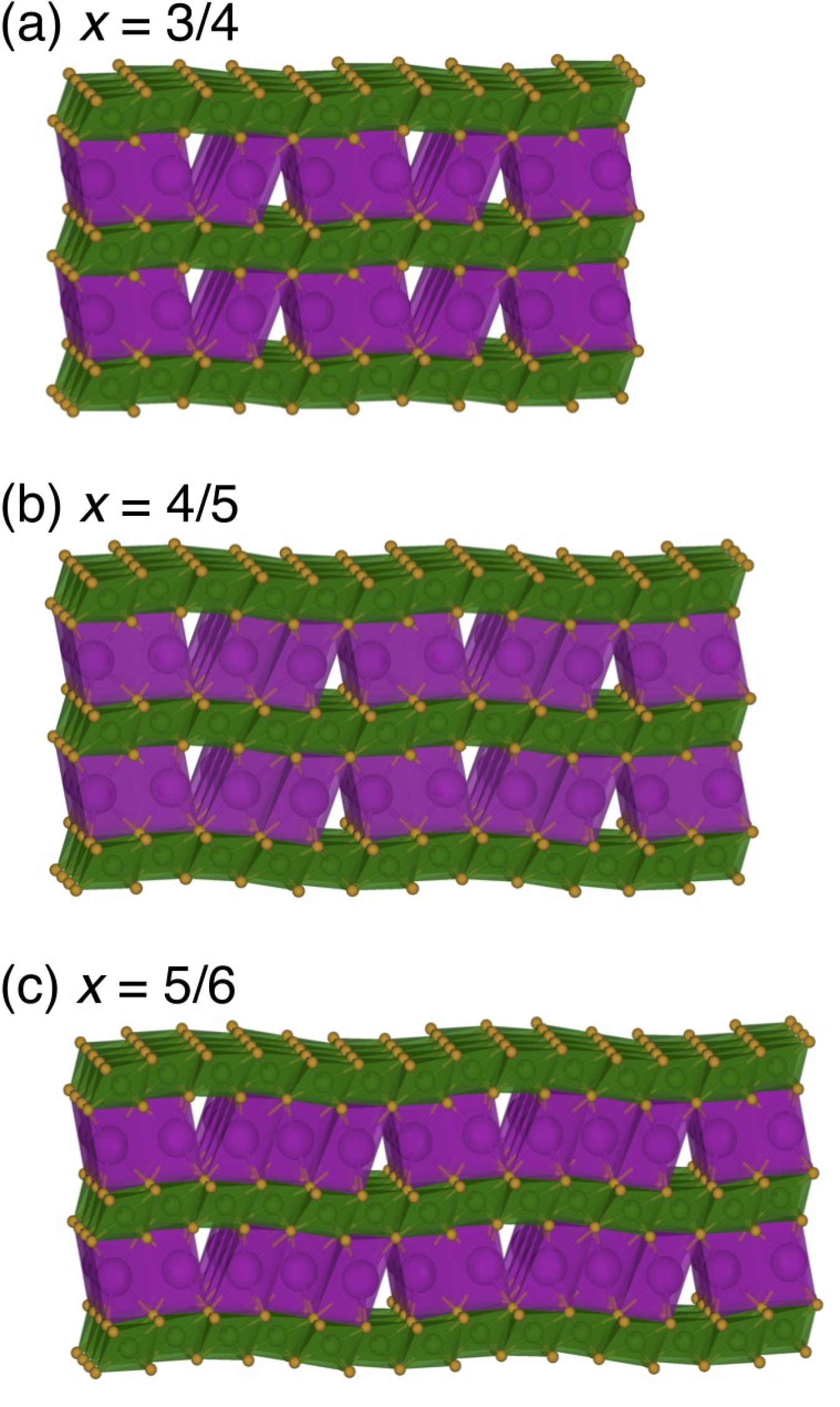
Predicted stable structures of layered potassium chromium oxide at varying potassium concentrations x. The structures belong to a staircase of orderings with varying proportions of prismatically and octahedrally coordinated potassium ions. Credit: Jonas L. Kaufman and Anton Van der Ven. “Ordering and Structural Transformations in Layered KxCrO2 for K-Ion Batteries.” Chemistry of Materials 32.15 (2020): 6392-6400.
“To do Monte Carlo, you need very large structures with thousands and thousands of atoms – larger than you would want to run in DFT,” Kaufman says. “You also need to evaluate many of them to get good averages” – also infeasible for DFT.
In one project, Kaufman and Van der Ven modeled the evolution of layered oxide materials used as sodium- and potassium-ion battery cathodes. These compounds frequently fall into strict repetitive structures inside the layered oxide host and change phases, causing large voltage steps, degrading the materials and sapping the battery’s power-producing capability.
The models varied the sodium or potassium content inserted into layered cobalt oxide. “We predicted this so-called devil’s staircase, where you have many ordered phases right next to one another” rather than a single main transition. The small steps between neighboring phases produce the sloping voltage graph. The results agree well with experiments, Kaufman says. The researchers published related work on potassium chromium oxide in Chemistry of Materials in 2020.
Michael Toriyama, then a University of Illinois at Urbana-Champaign undergraduate, contributed to the potassium cobalt oxide work while on a summer research project. He’s now a DOE CSGF recipient at Northwestern University.
Most of Kaufman’s calculations are on a local cluster at his university, but he’s also used the Cori supercomputer at DOE’s National Energy Research Scientific Computing Center.
In a switch from mostly modeling static crystals, Kaufman calculated dynamic interactions on his 2019 Lawrence Livermore National Laboratory practicum. His supervisor, DOE CSGF alumnus Brandon Wood, helps lead the Hydrogen Materials – Advanced Research Consortium, a multi-lab effort to develop hydrogen storage compounds.
Kaufman learned ab initio (first principles) molecular dynamics (AIMD) techniques to model molecular interactions. He calculated thermodynamic properties for surfaces in metal hydrides, metals that bond with and may store hydrogen. The goal: understand the materials’ performance at the nanoscale, where surface effects are important.
Kaufman’s simulations were successful, but the practicum ended before he could fully analyze the results. He’s continued the collaboration with the Livermore researchers.
Meanwhile, Kaufman did a second project: modeling how hydrogen releases from non-crystalline metal hydrides, a proxy for processes at interfaces between materials. He initially found the materials’ disorder and low densities may produce hydrogen release at lower temperatures than in crystalline materials – an important storage-material property. Follow-up calculations, however, found the effect probably wasn’t real. Nonetheless, it was a learning experience, Kaufman says, and he’s now considering incorporating AIMD into his doctoral research.
His practicum also has led Kaufman to consider seeking laboratory positions after 2022 graduation. Meanwhile, he still meets with Pribram-Jones and Bassman’s group – becoming an ad hoc alumni advisor on DFT and other techniques.